Pear
Pear is a tool to merge paired-end sequencing reads, prior to downstream tasks such as assembly.
Get data
Input: paired-end reads.
- We will use a set of Illumina MiSeq reads from the bacteria Staphylococcus aureus.
Go to your Galaxy server.
- In the tool panel, go to
Get Data: Upload File - Select
Paste/Fetch data - In the box, paste in:
- Click
Start and thenClose . - These two files will upload to your current Galaxy history.
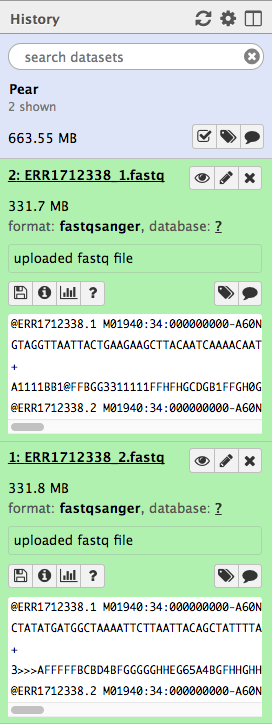
- Using the pencil icon, change the filetype to “fastqsanger”, and shorten the name of the file.
Run Pear
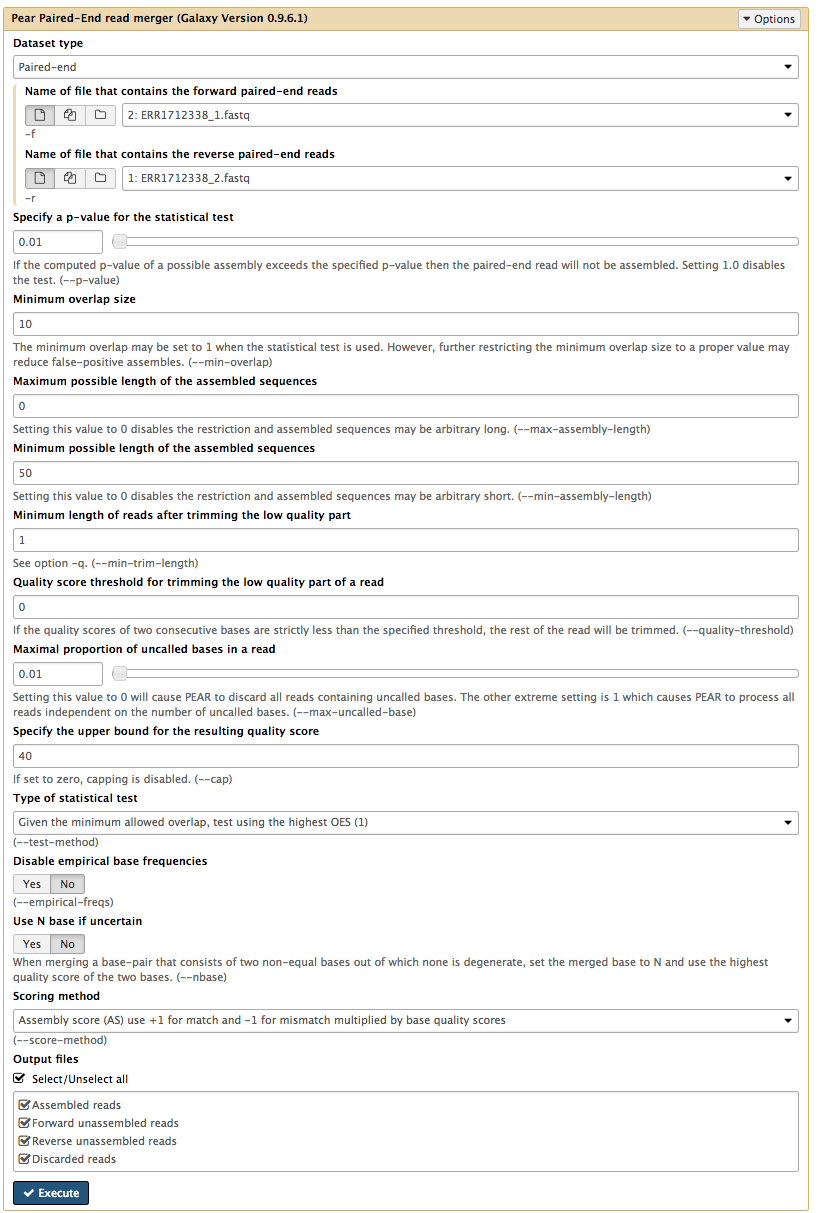
In the tool panel, go to
Dataset type : Paired-endName of file that contains the forward paired-end reads :ERR1712338_1.fastq Name of file that contains the reverse paired-end reads :ERR1712338_2.fastq - Leave other settings as per defaults, except:
Maximal proportion of uncalled bases in a read : 0.01- omits reads if >1% of the reads is missing (N)
Output files : Select all
Your tool interface should look like this:
- Click
Execute
Results
There are four output files.
Assembled reads : merged paired-end reads.Unassembled forward reads andUnassembled reverse reads : remaining, unmerged reads.Discarded reads : Did not meet quality specified
In this case, most of the reads have been merged (~360MB); 90MB are unmerged, and 350 sequences have been discarded.
Next
Run Trimmomatic to trim sequences before assembling.